3 靶向G蛋白偶聯(lián)受體的肽類藥物及其發(fā)現(xiàn)
人體內(nèi)有多種CPCR的內(nèi)源性多肽類配體,例如降鈣素,催產(chǎn)素����、生長抑素���、胰高血糖素樣肽�����、加壓素�����、甲狀旁腺素和促性腺激素釋放激素等,對這些多肽的改造和修飾是多肽藥物開發(fā)的主要方向之�。目前,上市的將近50種靶向GPCR的肽類藥物的主要靶點包括胰高血糖素樣肽受體�����、甲狀旁腺素受體、生長抑素受體�����、促性腺激素釋放激素受體�����、加壓素/催產(chǎn)素受體和鈣敏感受體等,用于治療代謝性疾病��、神經(jīng)系統(tǒng)疾病和癌癥等多種疾病���。已上市的多肽藥物多是通過化學(xué)合成(例如固相多肽合成法) ,或者基因重組技術(shù)合成的擁有與內(nèi)源性多肽配體相似序列的多肽類似物及其衍生物。
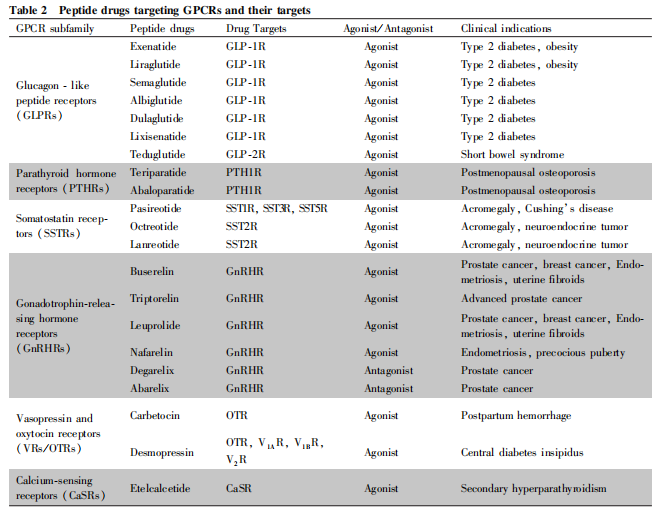
下文將重點介紹幾類研發(fā)成功已上市的靶向GPCR肽類藥物(見Table 2 )���。
3.1 靶向胰高血糖素樣肽1受體的肽類藥物
胰高血糖素樣肽-1受體( glucagon-like peptide-1receptor��,GLP-1R)主要在胰腺中表達,其激動劑類藥物是近年來降糖藥物研發(fā)的熱點����。CLP-1R的天然多肽類配體胰高血糖素樣肽1(glucagon-like pep-tide-1�,GLP-1)是37個氨基酸的肽�����。攝入葡萄糖后,腸道內(nèi)分泌L細胞合成并分泌CLP-1 ,作用于CLP-1R,引起細胞內(nèi)cAMP增加,促進葡萄糖誘導(dǎo)的胰島素分泌,抑制胰高血糖素的釋放���。除了其特有的葡萄糖依賴的胰腺作用外,GLP-1還發(fā)揮著降低食欲和促進飽腹感的中樞作用,突出了其作為糖尿病或肥胖癥療法的潛。天然GLP-1半衰期短,會被體內(nèi)二肽基肽酶Ⅳ ( dipeptidyl pepti-dase IV,DPP-Ⅳ)迅速降解并被腎清除,不適合直接作為藥物應(yīng)用�。目前,已有7種多肽類GLP-1R激動劑上市,用于治療2型糖尿病。
3.1.1 Exendin-4及其衍生物
1992年,Eng 等從Gila毒蜥的毒液中分離出一種天然肽Exendin-4,具有與GLP-1相似的藥理學(xué)特性��。例如,可增加胰島素分泌����、降低血糖水平等。與GLP-1不同的是,Exendin-4因其N-端第2位的丙氨酸替換為甘氨酸,不被DPP-IV降解,因此具有更長的半衰期和較強的生物活性�。Exenatide(艾塞那肽)即人工合成的Exendin-4,包含39個氨基酸,在2005年獲得FDA批準(zhǔn),是第1個用于治療⒉型糖尿病的GPCR靶向藥。Exenatide可以與CLP-1R結(jié)合改變其構(gòu)象,激活A(yù)C,使細胞中cAMP增加,從而促進葡萄糖依賴的胰島素分泌���、抑制胰高血糖素分泌和增加胰島素敏感性,達到降低血糖的目的���。
目前, Exenatide有每日2次(Exenatide BID)和每周1次(Exenatide QW)兩種劑型。Exenatide BID除可有效降低患者的血糖外,還能發(fā)揮降低體重的作用,特別適合作為合并向心性肥胖的2型糖尿病患者的治療藥物�。但由于Exenatide BID 需要頻繁皮下注射,可能導(dǎo)致患者依從性差。Exenatide QW是首個實現(xiàn)一周1次給藥的GLP-1R激動劑,其通過微球技術(shù),使Exenatide在體內(nèi)緩慢釋放,發(fā)揮長效降糖作用。繼Exenatide QW后,人們也采用了多種其他策略來提高GLP-1R激動劑的血漿穩(wěn)定性����,以延長其半衰期。這些策略可大致分為兩種:一種是對Exenatide 的序列進行修飾,延長其血漿半衰期,例如Lixisenatide(利西拉肽) ,省略了肽 Ex-enatide序列中第38位的脯氨酸并在其C-端添加了6個賴氨酸殘基和1個酰胺基,這些修飾延長了其血漿半衰期��。
3.1.2 胰高血糖素樣肽1衍生物
另一種策略則側(cè)重于修飾天然的GLP-1,例如Liraglutide(利拉魯肽)�、Albiglutide(阿必魯肽)、 Dulaglutide(度拉魯肽)和Semaglutide(索馬魯肽)���。Liraglutide于2010年獲得FDA批準(zhǔn),其將天然CLP-1上的第34位賴氨酸替換為精氨酸,并在第26位賴氨酸上增加了1個連接谷氨酸的C16棕櫚脂肪酸側(cè)鏈,從而增加了其與血漿白蛋白的結(jié)合,降低了DPP-Ⅳ的酶切作用和腎的清除速率��。除以與 Exenatide類似的機制發(fā)揮降糖作用外, Liraglutide 與CLP-IR結(jié)合后還能延緩胃排空,在治療肥胖癥方面效果顯著。Albig-lutide和l Dulaglutide均是以融合蛋白質(zhì)形式生產(chǎn)出來的,Albiglutide是通過基因重組技術(shù)利用酵母產(chǎn)生的,與人血清白蛋白融合,其將天然CLP-1 上第2位的丙氨酸替換為甘氨酸以抵抗DPP-Ⅳ降解:; Dulaglutide是利用哺乳動物細胞培養(yǎng)產(chǎn)生的�����,共價連接到人IgG4-Fc重鏈上,這些修飾分別降低了這2種藥物腎的清除率,增加了其藥理活性的持續(xù)時間���。
與上述幾種GLP-1R 激動劑相比, Semaglutide最主要的突破是實現(xiàn)了CLP-1R激動劑的長效制劑和口服制劑��。2017年, Semaglutide注射劑被FDA批準(zhǔn)上市,可延長到每周注射1次�����。其長效機制是基于對結(jié)構(gòu)的化學(xué)修飾���。在 Semaglutide 中,天然GLP-1的第8�、34位氨基酸分別被2-氨基異丁酸(2-aminoisobutyric acid)和精氨酸取代,使其能有效避免被 DPP-IV降解;并且,其中天然CLP-1的第26位賴氨酸被硬脂酸?;? acylated with stearic diac-id) ,能夠使其與人血清白蛋白結(jié)合,從而延長血漿半衰期并避免被腎快速清除。這些修飾都延長了多肽在體內(nèi)循環(huán)的時間,使其半衰期可以達到7d之久�����。Semaglutide口服制劑剛于2019年被FDA批準(zhǔn)上市,其利用了Eligen公司研發(fā)的基于促吸收劑的大分子遞送技術(shù):大分子藥物被多個促吸收劑SNAC[ 8-(2-羥基苯甲酰胺基)辛酸鈉]分子包裹形成脂質(zhì)體結(jié)構(gòu),可保護多肽藥物不被胃中的酶類降解�。
3.2 靶向胰高血糖素樣肽2受體的肽類藥物
胰高血糖素樣肽2受體( glucagon-like peptide-2receptor ,GLP-2 R)主要分布于胃腸道組織中,通過cAMP依賴性信號轉(zhuǎn)導(dǎo)途徑實現(xiàn)對胃腸道的調(diào)控�����,增強腸道營養(yǎng)吸收�。GLP-2R的天然肽類配體胰高血糖素樣肽2( glucagon-like peptide-2.GLP-2)是由33個氨基酸組成的腸源性多肽,由腸道內(nèi)分泌L細胞分泌,可被 DPP-IV快速降解,半衰期非常短,僅為7min。 Teduglutide (替度魯肽)是一種由DNA重組技術(shù)合成的GLP-2類似物��,2012年被FDA批準(zhǔn)為治療依賴腸外營養(yǎng)支持的成人短腸綜合征的孤兒藥����。短腸綜合征( short bowel syndrome , sBS)是一種罕見的具有潛在生命危險的吸收不良疾病,其原因是先天性缺陷.疾病導(dǎo)致的吸收障礙或廣泛手術(shù)切除導(dǎo)致大部分腸道功能喪失。當(dāng)SBS患者腸道吸收營養(yǎng)物質(zhì)�、電解質(zhì)和水的能力不能滿足機體需要時,需要腸外營養(yǎng)支持。在Teduglutide 中 ,天然CLP-2的第2位丙氨酸被替換為甘氨酸以抵抗DPP-IV降解、延長血漿半衰期���,其可通過作用于GLP-2R來發(fā)揮生物學(xué)作用,實現(xiàn)對胃腸道的調(diào)控,減少胃排空和分泌,并促進小腸粘膜上皮細胞的生長�、增殖和修復(fù),從而增加小腸吸收��、減少腹瀉��。
3.3 靶向甲狀旁腺素1受體的肽類藥物
甲狀旁腺素Ⅰ受體( para thyroid hormone 1 re-ceptor ����,PTHIR)主要表達于腎和骨骼,存在2種不同的高親和性構(gòu)象:G蛋白非依賴構(gòu)象(RO)和G蛋白依賴構(gòu)象(RG)。與RO構(gòu)象親和力更高的配體主要激活β-抑制蛋白信號通路而引發(fā)受體內(nèi)吞,觸發(fā)長時程信號反應(yīng);與RG構(gòu)象親和力更高的配體主要激活G蛋白介導(dǎo)的cAMP依賴性信號轉(zhuǎn)導(dǎo)通路,觸發(fā)瞬時信號反�����。甲狀旁腺激素( parathy-roid homone��,PTH)和甲狀旁腺激素相關(guān)肽(PTH-related peptide�����,PTHrP)是骨代謝過程中重要的調(diào)節(jié)因子,甲狀旁腺激素相關(guān)肽的N-末端含有甲狀旁腺激素同源序列,二者均可作用于PTHIR通過cAMP依賴性的PKA信號轉(zhuǎn)導(dǎo)通路調(diào)節(jié)骨代謝��。不同的是甲狀旁腺激素主要調(diào)節(jié)鈣的穩(wěn)態(tài)和骨吸收,而甲狀旁腺激素相關(guān)肽是促進骨形成的關(guān)鍵肽����。
3.3.1 甲狀旁腺激素類似物
Teriparatide Teripa-ratide(特立帕肽)是含有34個氨基酸的甲狀旁腺激素類似物,也是第1個被FDA批準(zhǔn)上市的調(diào)節(jié)骨代謝的肽類藥物,其主要作用于甲狀旁腺素Ⅰ受體的RO構(gòu)象,促進β-抑制蛋白的結(jié)合而導(dǎo)致內(nèi)吞過程,可持續(xù)調(diào)節(jié)cAMP含量,從而持續(xù)性激活下游信號通路,介導(dǎo)長時程信號轉(zhuǎn)導(dǎo),加快骨吸收�����。
3.3.2 甲狀旁腺激素相關(guān)肽類似物
AbaloparatideAbaloparatide(阿巴洛肽)是第1個被開發(fā)的甲狀旁腺激素相關(guān)肽類似物,具有34個氨基酸,于2017年被FDA批準(zhǔn)用于絕經(jīng)后女性骨質(zhì)疏松癥的治療�。其主要作用于甲狀旁腺素Ⅰ受體的RG構(gòu)象,引發(fā)瞬時的cAMP含量的增高,介導(dǎo)短暫的信號反應(yīng),促進骨形成的能力超過骨吸收。這2種藥物靶向同一受體甲狀旁腺素1受體,但由于其構(gòu)象偏好性存在差異,它們激活同一靶點的下游信號通路不同,導(dǎo)致Abaloparatide對于絕經(jīng)后女性骨質(zhì)疏松癥的療效優(yōu)于Teriparatidel��。這也啟示在研發(fā)靶向GPCR的肽類藥物時應(yīng)考慮其構(gòu)象偏好性�。
3.4 靶向生長抑素受體的肽類藥物
生長抑素受體( somatostatin receptor ,SSTR)家族包括SSTR1-5,廣泛分布于中樞神經(jīng)系統(tǒng)���、垂體和許多外周器官����。生長抑素受體的天然配體生長抑素( somatostatin�����,SST)與受體結(jié)合可誘導(dǎo)cAMP依賴性信號轉(zhuǎn)導(dǎo)途徑,抑制各種促腫瘤生長的激素和生長因子的釋放,從而抑制癌細胞增殖或誘導(dǎo)癌細胞凋亡����。生長抑素對受體具有很高的親和力,但在血漿中的半衰期非常短,僅為1~ 3 minl7]����。一種人工合成的天然生長抑素的八肽衍生物—-環(huán)狀生長抑素受體激動劑Octreotide (奧曲肽)可用于治療神經(jīng)內(nèi)分泌腫瘤和肢端肥大癥��。通過引入D-氨基酸,Octreotide 血漿半衰期可達72~113 min,并可選擇性地與生長抑素受體和生長抑素受體5結(jié)合��。Octreotide與生長抑素受體結(jié)合后,通過PLC介導(dǎo)的信號轉(zhuǎn)導(dǎo)通路,產(chǎn)生第二信使IP,并可激活L型Ca*通道,抑制生長激素的產(chǎn)生�����。Pasireotide(帕瑞肽)是另一種生長抑素受體激動劑,是美國和歐盟批準(zhǔn)的治療庫欣綜合征的孤兒藥�。Pa-sireotide對生長抑素受體5的親和力較高,能抑制促腎上腺皮質(zhì)激素( adreno cortico tropic hormone,ACTH)的分泌,使庫欣綜合征患者的皮質(zhì)醇分泌減少。
3.5 靶向促性腺激素釋放激素受體的肽類藥物
促性腺激素釋放激素受體( gonadotropin-relea-sing hormone receptor �����,GnRHR)主要在垂體和生殖系統(tǒng)相關(guān)的組織器官中表達,其天然肽類配體促性腺激素釋放激素( gonadotropin-releasing hormone,GnRH)是下丘腦分泌產(chǎn)生的一種十肽神經(jīng)激素,在生殖調(diào)控中發(fā)揮重要作用���。
3.5.1 促性腺激素釋放激素受體激動劑
Leuprolide促性腺激素釋放激素類似物L(fēng)euprolide(亮丙瑞林)是促性腺激素釋放激素受體激動劑, 1985年被FDA批準(zhǔn)上市,用于治療激素反應(yīng)性癌癥,例如前列腺癌和乳腺癌,也可用于治療其它雌激素依賴性疾病,例如子宮內(nèi)膜異位癥和子宮肌瘤�����。Leuprolide主要通過Gaqm與 GPCR相關(guān)聯(lián),激活PLC,釋放P和DAG,誘導(dǎo)PKC激活,促使垂體前葉釋放黃體生成素(luteinizing hormone,LH)和卵泡刺激素( follicle-stimulating hormone��,F(xiàn)SH),最終通過下丘腦-垂體-性腺軸的正常生理作用,誘導(dǎo)血清雌二醇和睪酮水平的一過性升高。而下丘腦-垂體-性腺軸的調(diào)節(jié)依賴于下丘腦促性腺激素釋放激素的脈沖分泌,導(dǎo)致Leuprolide 連續(xù)幾周治療后促性腺激素釋放激素受體敏感性下降�����。這種促性腺激素釋放激素受體活性的長期下調(diào)是Leuprolide治療的主要機制��,最終導(dǎo)致黃體生成素和卵泡刺激素分泌減少,性腺功能減退,從而使雌二醇和睪酮水平顯著降低,這一點與其他GPCR的激動劑不同�。
3.5.2 促性腺激素釋放激素受體拮抗劑
Degarelix在激素敏感型前列腺癌的治療中, Leuprolide所導(dǎo)致的睪酮水平的初始升高反而會使前列腺癌的癥狀加重,這促使研究者進一步開發(fā)了促性腺激素釋放激素受體拮抗劑。2008年,FDA批準(zhǔn)了一種促性腺激素釋放激素受體拮抗劑 Degarelix(地加瑞克) ,用于治療晚期前列腺癌�����。Degarelix 與促性腺激素釋放激素受體可逆性結(jié)合,下調(diào)細胞內(nèi)cAMP含量,通過抑制cAMP依賴的GPCR信號通路減少促性腺激素的釋放,進而減少睪酮的釋放以阻止前列腺癌的生長和惡化�。通過在肽鏈的第5,6位引入P-脲基苯丙氨酸( p-ureido-phenylalanines ) , Degarelix在實現(xiàn)藥物作用時間延長的同時也避免了因組胺釋放引起的超敏反應(yīng)。有研究表明,在治療晚期前列腺癌方面, Degarelix 優(yōu)于Leuprolidel�����。
3.6 靶向加壓素/催產(chǎn)素受體的肽類藥物
加壓素受體( vasopressin receptors�����,VRs )包括V,xR �����、 V,gR和 V,R。V,xR主要在肝���、血管平滑肌和血小板中表達,促進血管收縮和血小板聚集; VR主要在垂體中表達,可促進促腎上腺皮質(zhì)激素的釋放;V,R主要在腎集合管中表達,具有抗利尿作用���。催產(chǎn)素受體( oxytocin receptors,OTRs)主要分布于子宮和乳腺����。加壓素( vasopressin)和催產(chǎn)素( oxytocin)均為九肽,主要通過結(jié)合其受體來發(fā)揮作用, VnR 、VR與催產(chǎn)素受體作用機制相似,主要通過偶聯(lián)的Gαx.刺激PIC的活性,釋放P��。和DAG ,誘導(dǎo)內(nèi)質(zhì)網(wǎng)Ca*釋放;而V,R主要與Go.偶聯(lián),激活A(yù)C產(chǎn)生cAMP,誘導(dǎo)PKA激活��。Carbetocin(卡貝縮宮素)是人工修飾的催產(chǎn)素類似物,天然催產(chǎn)素酚羥基上的氫被甲基取代,半胱氨酸殘基上的氨基和硫分別被氫和亞甲基取代,這些修飾延長了Carbetocin 的作用時間��。Carbetocin可與子宮平滑肌催產(chǎn)素受體結(jié)合,誘導(dǎo)子宮節(jié)律性收縮,增加子宮收縮頻率和強度,適用于控制分娩后出血�。Desmopressin(去氨加壓素)是人工合成的九肽化合物,2017年被FDA批準(zhǔn)用于治療中樞性尿崩癥,其對V,R作用最強,可通過提高腎集合管上皮細胞cAMP水平促使腎血管舒張發(fā)揮抗利尿作用,因此與天然加壓素相比, Desmopressin抗利尿作用顯著增強,而對平滑肌的作用卻減弱,從而避免了高血壓引起的不良作用。
3.7 靶向鈣敏感受體的肽類藥物
鈣敏感受體( calcium-sensing receptor ����,CaSR ) 廣泛分布于甲狀旁腺、胃腸道����、腎、骨組織等參與調(diào)節(jié)體內(nèi)鈣穩(wěn)態(tài)的組織器官中�����。血鈣水平升高激活鈣敏感受體,從而抑制了甲狀旁腺激素的分泌;相反,血鈣水平的降低會使鈣敏感受體的活性受到抑制,促進甲狀旁腺激素的分泌����。繼發(fā)性甲狀旁腺功能亢進癥( secondary hyperparathy roidism,SHPT)是慢性腎疾病( chronic kidney disease ���,CKD)常見的一種慢性進行性并發(fā)癥����。SHPT的特征是甲狀旁腺增生引起的甲狀旁腺激素水平升高,以及鈣和磷等礦物質(zhì)代謝異常����。這些礦物質(zhì)失衡會導(dǎo)致嚴重的并發(fā)癥,例如骨骼疾病、軟組織鈣化����、血管鈣化和心血管并發(fā)癥等。Etelcalcetide(維拉卡肽)是鈣敏感受體的一種新型長效肽激動劑,2017年被FDA批準(zhǔn)用于治療成年血液透析患者繼發(fā)性甲狀旁腺功能亢進���。Etelca lcetide直接結(jié)合開激活中狀z陳上的鈣敏感受體,通過PLC將PPa紅群心多一福使IP,和 DAG,從而抑制甲狀旁腺中的甲狀旁腺激素分泌����。
4 問題與展望
肽類藥物以其生物活性尚.1低理、就王板沂特點成為了近年藥物研發(fā)的熱點,尤其是結(jié)構(gòu)上近乎無限的可能性使之更適合于靶向GPCR����。然而,噬菌體展示等技術(shù)在很大程度上依賴于活性重組蛋白質(zhì)的制備與固相化,目前尚不能對corkinas.通道��、酪氨酸激酶受體( receptor tyrosine kinase���,RTK)等跨膜蛋白質(zhì)進行篩選���。另外,基于親和力的篩選過程并不能區(qū)別跨膜蛋白質(zhì)的激動劑和拮抗劑,即不能進行功能篩選。
Lerner 課題組開發(fā)的自分泌信號系統(tǒng)首次實現(xiàn)了哺乳動物細胞中的肽類咫示皮切HE州主穿肽左較噬菌體展示技術(shù),此系統(tǒng)還不能女就E含NNK的篩選,原因在于該體系中肽庫豐度是來自含NNK簡并密碼子( degenerate codons )的PC房法成過質(zhì)粒構(gòu)建轉(zhuǎn)化提取����、質(zhì)粒轉(zhuǎn)染L內(nèi)干細晌染哺乳動物細胞等一系列操作后,最終傳手J在以表面的隨機肽庫豐度存在大幅下降(肽庫豐度在以上步驟中不斷丟失)。而肽庫豐度高低是篩選有效候選肽的關(guān)鍵因素��。因此,在Lerner 確立了哺乳動物細胞系肽類展示篩選技術(shù)之后,能否在哺乳動物細胞內(nèi)建立復(fù)雜肽庫是當(dāng)前肽類藥物開發(fā)的一大挑戰(zhàn)���。
我們課題組開發(fā)了一種基于線性雙鏈DNA( linear double-stranded DNA)的“與門(AND Gate)”邏輯基因線路設(shè)計新策略��。通過PCR反應(yīng)將完整的基因表達模塊(啟動子-基因編碼區(qū)-poly(A)尾信號)拆分為2個線性雙鏈 DNA分子引入哺乳動物細胞,其在細胞內(nèi)經(jīng)非同源末端連接(non-homologousend joining��,NHEJ) ,或同源重組( homologous recom-bination���,HR)機制連接在一起重新形成完整基因表達模塊,實現(xiàn)“與門”運算及基因表達(見Fig.2)。
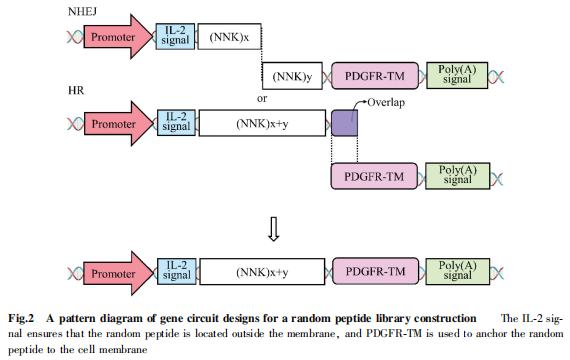
由此,我們建立了一種在哺乳動物細胞系內(nèi)引入高豐度肽庫的方法�����。隨著基于哺乳動物細胞系的高豐度肽庫構(gòu)建策略的開發(fā)及自分泌篩選體系的建立�����,靶向GPCR 的肽類藥物篩選體系將不斷完善并趨于成熟,這將為更多肽類藥物的開發(fā)建立堅實的基礎(chǔ)�。
相關(guān)鏈接:激素,促性腺激素���,胰高血糖素��,細胞���,北納生物










登錄后才可以評論